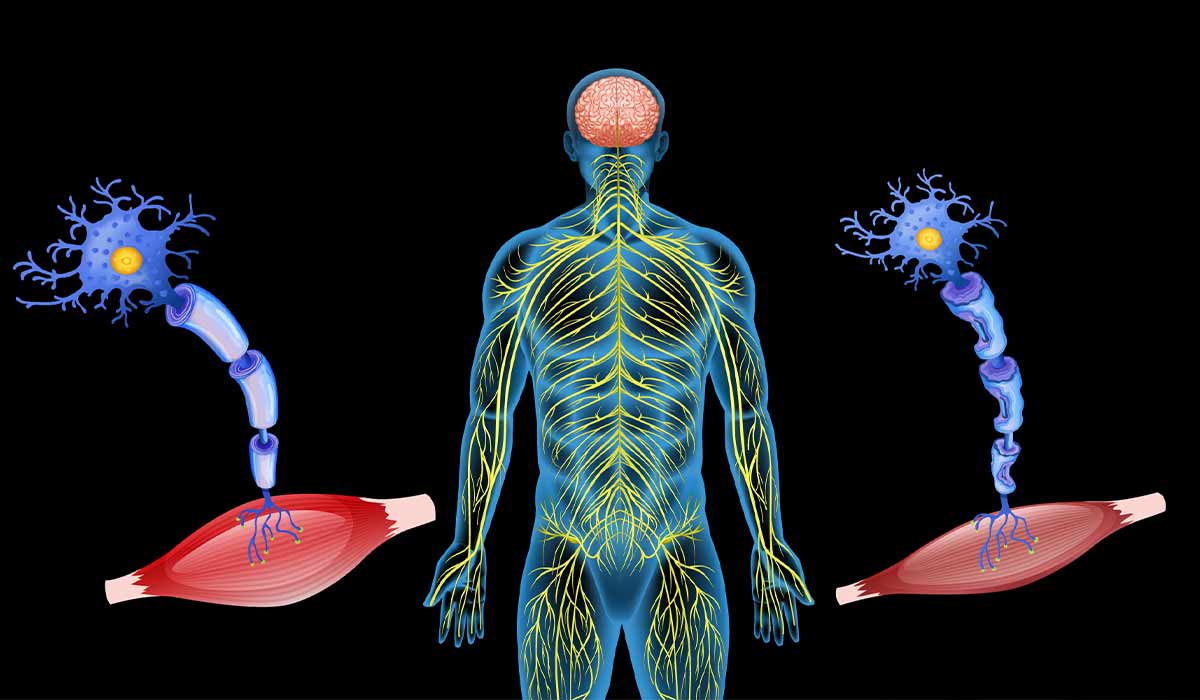
Amyotrophic Lateral Sclerosis is a severe disease that causes damage to the central or peripheral nervous system. It is characterized by gradual damage to nerve fibers responsible for the functioning of muscles and motor neurons in the brain.
There are three forms of ALS:
The type and severity of the first symptoms of amyotrophic lateral sclerosis is a very individual matter. The condition attacks the motor nerves, but mental performance remains intact. Patients are also doomed to pain exacerbated by stress, depression, and anxiety.
There is no known clear cause![]() of the development of ALS. Conclusions from previous studies indicate a multifactorial etiology related to a specific genetic predisposition
of the development of ALS. Conclusions from previous studies indicate a multifactorial etiology related to a specific genetic predisposition![]() . Factors increasing the risk of amyotrophic lateral sclerosis include childhood infections, toxins damaging the nervous system, and intense sports practice.
. Factors increasing the risk of amyotrophic lateral sclerosis include childhood infections, toxins damaging the nervous system, and intense sports practice.
In some ALS patients, it runs in families, and in such cases, genetic mutations have been identified that lead to the development of the disease.
The most common form of amyotrophic lateral sclerosis is the so-called Charcot-Marie-Tooth disease, which initially manifests itself with involuntary muscle tremors, weakness of the limbs, and painful cramps (limb form). It starts with the muscles of the hands, progresses towards the forearms, then affects the arms. These symptoms are usually asymmetric. Typically, the disease progresses slowly and worsens over many years.
The first symptoms of amyotrophic lateral sclerosis vary in intensity and location in individual patients. Rarely do the initial signs of ALS affect the muscles of the shins and feet or only the arms muscles and shoulder girdle.
It happens that the disease first attacks the muscles of the jaw and tongue, which causes speech disorders and difficulties in chewing and swallowing food (a form with features of bulbar syndrome). You can then observe quite characteristic, involuntary “tremors” of the tongue, called fasciculations – a symptom of muscle fiber atrophy. Some patients develop pseudobulbar syndrome![]() – brain damage causes emotional lability, manifested by uncontrolled outbursts of laughter or crying unrelated to the situation.
– brain damage causes emotional lability, manifested by uncontrolled outbursts of laughter or crying unrelated to the situation.
The least common form of the disease is the form of breathing problems caused by the involvement of the intercostal muscles.
The characteristic involvement of the upper and lower motor neurons causes limb paresis and increased muscle tension – this is associated with spasticity, exaggerated reflexes, and the Babinski sign![]() . It involves pathological bending of the big toe towards the back of the foot when irritating its plantar surface. However, this is not characteristic of amyotrophic lateral sclerosis but indicates involvement of the upper motor neuron. Symptoms of lower neuron involvement include muscle atrophy, muscle mass loss, muscle fiber fasciculation, and muscle weakness.
. It involves pathological bending of the big toe towards the back of the foot when irritating its plantar surface. However, this is not characteristic of amyotrophic lateral sclerosis but indicates involvement of the upper motor neuron. Symptoms of lower neuron involvement include muscle atrophy, muscle mass loss, muscle fiber fasciculation, and muscle weakness.
If the described symptoms appear, you should see a primary care physician who, if necessary, will refer the patient to a neurologist for further diagnosis and treatment. Please remember that not all people who experience the above symptoms have amyotrophic lateral sclerosis. It is essential to establish the correct diagnosis early to avoid other diseases with similar manifestations that can be treated or have a relatively good prognosis.
Depending on the dominant symptoms, the differential diagnosis of ALS![]() includes diseases such as:
includes diseases such as:
The weakening of the muscular apparatus characterizes all these diseases. It is worth emphasizing that patients remain mentally fit until the end, which is often an additional burden for them – they see how the condition is progressing and are aware of their limitations and that ALS inevitably leads to death.
Multiple sclerosis![]() is often confused with amyotrophic lateral sclerosis. These are two different disease entities. Both diseases affect the nervous system. However, in the case of multiple sclerosis, nerve sheaths are destroyed. In the case of ALS, peripheral nerves that transmit impulses to the muscles are destroyed.
is often confused with amyotrophic lateral sclerosis. These are two different disease entities. Both diseases affect the nervous system. However, in the case of multiple sclerosis, nerve sheaths are destroyed. In the case of ALS, peripheral nerves that transmit impulses to the muscles are destroyed.
The difference between these two diseases is also the prognosis. These are much better for multiple sclerosis. Amyotrophic lateral sclerosis is characterized by a more severe course and a shorter life expectancy of patients.
The most important factors to establish the diagnosis![]() are the interview, i.e., information about the symptoms obtained from the patient, and a thorough neurological examination. After collecting a detailed history, the doctor will examine the patient, particularly on neurological assessment. During a neurological exam, the doctor assesses muscle strength (face and limbs) and muscle tone, looks at reflexes using a neurological hammer, and observes muscles for atrophy and fasciculation.
are the interview, i.e., information about the symptoms obtained from the patient, and a thorough neurological examination. After collecting a detailed history, the doctor will examine the patient, particularly on neurological assessment. During a neurological exam, the doctor assesses muscle strength (face and limbs) and muscle tone, looks at reflexes using a neurological hammer, and observes muscles for atrophy and fasciculation.
Theoretically, in some cases, amyotrophic lateral sclerosis can be diagnosed based on the interview and neurological examination alone. However, these are rare and usually occur when the disease is already advanced. Establishing the diagnosis at an early condition stage may not be easy and requires additional tests. They include laboratory blood tests, imaging tests, i.e., computed tomography or magnetic resonance imaging of the head or a specific section of the spine – depending on the symptoms – and electrophysiological examination (EMG).
Sometimes, as part of the diagnosis of amyotrophic lateral sclerosis, a lumbar puncture![]() is also performed to examine the cerebrospinal fluid. Laboratory blood tests, cerebrospinal fluid tests, and imaging tests are selected individually for each patient and are performed to exclude other causes of symptoms. Among all additional tests, the EMG test plays the greatest role in diagnosing amyotrophic lateral sclerosis. This test assesses the function of peripheral nerves and muscles.
is also performed to examine the cerebrospinal fluid. Laboratory blood tests, cerebrospinal fluid tests, and imaging tests are selected individually for each patient and are performed to exclude other causes of symptoms. Among all additional tests, the EMG test plays the greatest role in diagnosing amyotrophic lateral sclerosis. This test assesses the function of peripheral nerves and muscles.
It consists of two parts: a nerve conduction test, which involves stimulating peripheral nerves with an electrical stimulus and assessing their ability to conduct, and an electromyographic test, which involves recording the bioelectric activity of the muscle using a thin needle electrode inserted into it. This test allows us to confirm amyotrophic lateral sclerosis or to indicate other diseases of the neuromuscular system that cause similar symptoms.
Amyotrophic lateral sclerosis is an incurable![]() and progressive disease. There are no known effective methods for regaining fitness or even stopping the condition.
and progressive disease. There are no known effective methods for regaining fitness or even stopping the condition.

So far, there is no effective causal treatment for ALS; therapy focuses on alleviating the most troublesome symptoms of the disease. The only drug that extends the average lifespan of patients by about three months and delays the onset of respiratory failure is rulizone![]() – it is responsible for inhibiting the release of glutamate, which prevents damage to neurons.
– it is responsible for inhibiting the release of glutamate, which prevents damage to neurons.
Other medications used to treat ALS include medications to reduce muscle tension and reduce excessive salivation, medications to help the patient fall asleep and stabilize your mood, and painkillers. Psychotherapeutic support and speech therapy rehabilitation are also of great importance in ALS therapy. When breathing disorders occur, non-invasive ventilation methods using a respirator are used.
In the initial stage of the disease, all kinds of gymnastics and rehabilitation are necessary. Exercise helps extend the functioning of muscles and prevent stiffness and degeneration of joints. In addition, muscle spasticity (increased muscle tension) is reduced, eliminating pain.
Exercises must be tailored to the current individual capabilities of the patient, their age, and the severity of the disease. Speech therapy![]() is necessary for speech disorders. If such ailments occur, it is worth considering electronic communication devices, thanks to which the patient can communicate with the world (by printing a message from a machine).
is necessary for speech disorders. If such ailments occur, it is worth considering electronic communication devices, thanks to which the patient can communicate with the world (by printing a message from a machine).
It is recommended to install a so-called percutaneous endoscopic gastrostomy![]() (PEG). It is a drain placed directly into the stomach through an incision on the abdomen. Inserting a PEG reduces the risk of aspiration and allows for better nutrition of patients.
(PEG). It is a drain placed directly into the stomach through an incision on the abdomen. Inserting a PEG reduces the risk of aspiration and allows for better nutrition of patients.
However, in patients who experience breathing problems, non-invasive ventilation is recommended. It supports breathing using a respirator through a mask placed on the patient's face. Such ventilation does not occur continuously but in sessions – for example, one hour during the day and several hours at night (the duration of ventilation depends on the needs of the individual patient).
Moreover, it is worth including the family in the treatment and education process, whose support, daily help, and care are critical.
In difficult life situations, when you cannot take care of your loved ones yourself, it is worth seeking care in palliative medicine centers![]() . The patient will be provided with high-quality medical care, as well as rehabilitation and occupational therapy.
. The patient will be provided with high-quality medical care, as well as rehabilitation and occupational therapy.
Maintaining proper body weight is crucial for the correct functioning of the body and the fight against disease. It is the main purpose of the proceedings. Malnutrition is defined as a BMI (Body Mass Index) below 18.5 or a weight loss of more than 10% during the disease. It has been proven that a higher BMI![]() leads to a longer survival time in ALS patients. Similarly, low BMI leads to a more aggressive course of the disease.
leads to a longer survival time in ALS patients. Similarly, low BMI leads to a more aggressive course of the disease.
Research shows that thicker adipose tissue in a patient's premorbid period reduces the risk of ALS-related death. Fortunately, a well-selected diet![]() can compensate for the decreasing BMI and supplement the patient's increased energy needs. During the disease, it is recommended to check the patient's body weight periodically. The use of any slimming diet is contraindicated, even in overweight patients.
can compensate for the decreasing BMI and supplement the patient's increased energy needs. During the disease, it is recommended to check the patient's body weight periodically. The use of any slimming diet is contraindicated, even in overweight patients.
There is no one ideal diet that is suitable for all ALS patients. It is not entirely clear which nutrients are most beneficial for patients – while some individuals lean towards consuming large amounts of carbohydrates, others suggest a diet high in fat (ketogenic diet). However, the most significant thing is preventing malnutrition, including increasing the amount of calories consumed. It is estimated that the energy demand of a sick person may be approximately 15% higher than that of healthy people. Therefore, the average diet consumed in the period before the disease will not cover the amount of necessary calories.
To slow down the process of muscle degeneration and maintain the proper functioning of the immune system, the diet should be balanced, covering all groups of food products (unless there are allergies to individual ingredients), including cereal products, vegetables and fruits, dairy products, and fats. There are no foods contraindicated in the disease. Some studies suggest that pork, pork luncheon meats, and milk may hurt the course of the condition, as may alcohol, which may lead to interactions with other drugs. Additionally, you can supplement zinc, vitamin E and B12, L-carnitine, creatine, and folic acid. An appropriate diet helps improve the patient's well-being and quality of life.

After making the diagnosis, it is necessary to constantly monitor the patient's deteriorating swallowing and chewing functions, which may discourage them from taking sufficient amounts of food. Methods that can help improve the comfort of eating in patients with dysphagia and weakness of the masticatory muscles include:
Loss of appetite is usually observed in ALS patients, so it is worth following some tips that can support proper nutrition::
As the disease progresses, the patient's nutrition becomes more and more demanding and often requires the introduction of various modifications. Therefore, it is worth it for the sick person not to be left alone with the above challenge but to have constant support from specialists such as a dietician and speech therapist who will determine the optimal treatment and diet at the right time. A team consisting of a neurologist, a gastroenterologist, a nurse, and the patient's closest caregivers should also be involved in the care.
When, despite the above tips, eating becomes very difficult or impossible, proper nutrition and sufficient calories may be administered through a gastric tube or percutaneous gastrostomy. Their insertion does not mean a complete inability to eat orally but rather a supportive way of eating while carefully feeding. As the disease progresses, it also provides psychological comfort by eliminating the patient's fear of possible choking and aspiration. To sum up, the gastric tube improves the patient's quality of life and ensures the intake of appropriate amounts of calories. Consequently, as scientifically confirmed, early placement of a feeding tube in patients with dysphagia prolongs their survival time.
Despite many studies on the pathomechanisms and possibilities related to new treatment methods, ALS remains an incurable disease with a progressive course.
The prognosis![]() for amyotrophic lateral sclerosis is very poor. In most patients, death usually occurs 3 to 5 years after diagnosis. The main cause is paralysis of the respiratory muscles or other complications of the disease, including infections.
for amyotrophic lateral sclerosis is very poor. In most patients, death usually occurs 3 to 5 years after diagnosis. The main cause is paralysis of the respiratory muscles or other complications of the disease, including infections.